Pulmonary Arterial Hypertension (PAH) is a rare blood vessel disorder of the lung in which the pressure in the pulmonary artery (the blood vessel that leads from the heart to the lungs) rises above normal levels and may become life threatening.
In the advanced stage of the disease, the blood vessels in the lungs are narrowed, thickened and stiff, causing difficulty for the heart to pump blood through, causing the heart muscle to weaken over time. Clots can form and block part of the vessel's lumen as well.
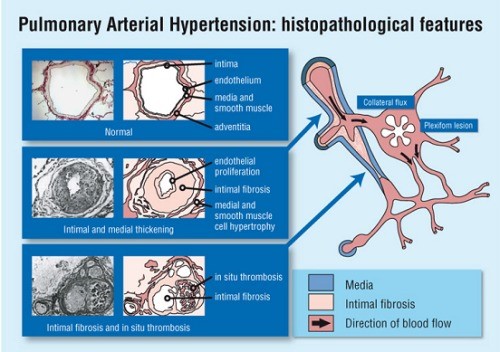
When PAH occurs in the absence of a known cause, it is referred to as Idiopathic Pulmonary Arterial Hypertension (IPAH). IPAH is extremely rare, occurring in about 2 to 10 persons per a million population per year. Causes of PAH include collagen vascular diseases (e.g. scleroderma, CREST syndrome or systemic lupus erythematosus), congenital heart diseases (shunts like ventricular and atrial septal defects), HIV infection, liver disease and diet drugs like fenfluramine and dexfenfluramine.
Common causes of secondary PAH include lung disorders (emphysema, lung fibrosis, sleep apnea), chronic pulmonary thromboembolism (chronic formation of blood clots in the pulmonary artery), and left heart disease (e.g. mitral stenosis).